Нам важлива ваша думка!
Відгук кожного пацієнта — це вклад у формування чесного рейтингу Довіри про медицину України

діюча речовина: рисдиплам (risdiplam);
1 пляшка має у складі 60 мг рисдипламу;
1 мл відновленого розчину має у складі 0,75 мг рисдипламу;
додаткові компоненти: маніт (E 421); ізомальт (Е 953); полуничний ароматизатор; кислота винна; натрію бензоат (E 211); поліетиленгліколь 6000; сахаралоза; кислота аскорбінова; динатрію едетат, дигідрат.
Порошок для орального розчину.
Основні фізико-хімічні властивості: порошок або порошок з грудочками або порошкова маса світло-жовтого або жовтого, або сірувато-жовтого, або зеленувато-жовтого, або світло-зеленого кольору. Відновлений розчин від зеленувато-жовтого до жовтого кольору.
Засоби, що впливають на опорно-руховий апарат. Інші засоби, що застосовуються за умови патології опорно-рухового апарату.
Код АТХ M09A X10.
Фармакодинаміка.
Механізм дії
Рисдиплам є модифікатором сплайсингу попередника матричної РНК (пре-мРНК) гена виживання мотонейронів 2 (SMN2), розробленим для терапія СМА, спричиненої мутаціями хромосоми 5q, що призводять до дефіциту білка SMN. Функціональний дефіцит білка SMN є патофізіологічним механізмом СMA усіх типів. Рисдиплам коригує сплайсинг SMN2, змінюючи баланс з виключення екзону 7 на включення екзону 7 в транскрипт мРНК, у результаті чого збільшується вироблення функціонального та стабільного білка SMN. Таким чином, рисдиплам лікує СМА завдяки підвищенню та утриманню функціонального рівня білка SMN.
Рисдиплам рівномірно розподіляється по всіх частинах тіла, у тому числі в центральну нервову систему, проникаючи через гематоенцефалічний бар’єр, і таким чином збільшуючи рівень білка SMN в ЦНС та усьому організмі. Концентрація рисдипламу в плазмі крові та білка SMN в крові відображає розподіл та фармакодинамічні ефекти рисдипламу в таких тканинах, як головний мозок та м’язи.
В клінічних дослідженнях FIREFISH, SUNFISH та JEWELFISH у хворих із інфантильним початком спінальної м’язової атрофії (СМА) та у хворих із пізнім початком СМА рисдиплам призводив до послідовного та тривалого збільшення визначеного у крові рівня білка SMN із зміною медіани більше ніж удвічі від вихідного рівня протягом 4 тижнів після початку терапія. Таке збільшення рівня білка SMN зберігалось протягом усього терапія впродовж щонайменше 24 місяців (див. підрозділ «Клінічна ефективність»).
Клінічна ефективність
Ефективність лікарський засібу Еврісді® у лікуванні хворих з інфантильною формою СMA (СМА 1 типу) та із формою СМА з пізнім початком (СМА 2 і 3 типу) була оцінена в 2 базових клінічних дослідженнях, FIREFISH та SUNFISH, і підтверджена додатковими даними з дослідження JEWELFISH. Ефективність лікарський засібу Еврісді® у лікуванні хворих із пресимптоматичною СМА була оцінена на підставі проміжного аналізу вторинних кінцевих точок дослідження RAINBOWFISH, що триває.
Пацієнти з клінічним діагнозом СМА 4 типу не брали участі у клінічних дослідженнях.
В клінічних дослідженнях була продемонстрована довгострокова ефективність впродовж щонайменше 24 місяців терапія. Існують обмежені дані використання лікарський засібу Еврісді® протягом більше 2 років.
Інфантильна форма СMA
Дослідження BP39056 (FIREFISH) – відкрите дослідження, що містить 2 частин, з вивчення ефективності, безпеки, фармакокінетики та фармакодинаміки лікарський засібу Еврісді® за участю хворих із СМА 1 типу та наявними симптомами (усі пацієнти мали генетично підтверджене захворювання із наявними 2 копіями гена SMN2). Частина 1 дослідження FIREFISH була розроблена як частина дослідження з пошуку дози. У підтверджувальній частині 2 дослідження FIREFISH оцінювалась ефективність лікарський засібу Еврісді® в терапевтичних дозах, обраних на підставі результатів, отриманих в частині 1 (див. розділ «Спосіб використання та дози»). Пацієнти з частини 1 не брали участі в частині 2.
Загалом 62 пацієнта із СМА 1 типу та наявними симптомами були включені в частину 1 (n = 21) і частину 2 (n = 41) дослідження FIREFISH, з яких 58 хворих отримували терапевтичну дозу лікарського засобу Еврісді®. Середній вік на момент появи клінічних симптомів становив 1,5 місяця (0,9–3 місяці). Середній вік на момент включення в дослідження становив 5,6 місяця (2,2–6,9 місяця), а середній час між появою симптомів і отриманням першої дози становив 3,7 місяця (1–6 місяців). 60 % хворих були жіночої статі, 57 % — європеоїдної раси і 29 % — азіатами. На початку дослідження медіанний бал CHOP-INTEND становив 23 (8–37) і медіанний бал HINE-2 становив 1 (0–5). Вихідні демографічні характеристики та характеристики захворювання хворих, включених в частину 1 дослідження, були співставні з такими для частини 2 дослідження.
Первинною кінцевою точкою була частка хворих, здатних сидіти без підтримки протягом щонайменше 5 секунд, як визначено у пункті 22 шкали розвитку немовлят та дітей раннього віку Бейлі — ІІІ видання (BSID-III) для оцінки загальної моторики, через 12 місяців терапія лікарський засібом Еврісді® в частині 2 дослідження; і такий результат був досягнутий у 29 % хворих (n = 12/41, 90 % ДІ: 17,8%, 43,1%, p <0,0001).
Ключові результати ефективності у хворих, які отримували терапія лікарський засібом Еврісді® в дослідженні FIREFISH (зведені дані, отримані в частині 1 і частині 2), наведені в таблиці 1.
Tаблиця 1
Резюме ключових результатів ефективності через 12 і 24 місяці (FIREFISH, частина 1 і частина 2)
| Кінцеві точки ефективності | Через 12 місяців | Через 24 місяці |
|
| Частка хворих (90 % ДІ) | |
| Етапи моторного розвитку та рухова функція | N = 58a | |
| BSID-III: сидіння без підтримки протягом щонайменше 5 секунд | 32,8 % (22,6 %, 44,3 %) | 60,3 % (48,7 %, 71,2 %) |
| CHOP-INTEND: кількість балів 40 або вище | 56,9 % (45,3 %, 68 %) | 74,1 % (63 %, 83,3 %) |
| CHOP-INTEND: збільшення на ≥ 4 бали від вихідного рівня | 89,7 % (80,6 %, 95,4 %) | 87,9 % (78,5 %, 94,2 %) |
| HINE-2: пацієнти, які відповіли відповідно до критеріїв розвитку моторної функціїb | 77,6 % (66,7 %, 86,2 %) | 82,8 % (72,5 %, 90,3 %) |
| Виживаність та виживаність без подій | N=62a | |
| Виживаність без подійc | 87,1 % (78,1 %, 92,6 %) | 83.8 % (74.3 %, 90.1 %) |
| Виживаність | 91,9 % (83,9 %, 96,1 %) | 90,3 % (81,9 %, 94,9 %) |
| Годування | N = 58a | |
| Здатність отримувати пероральне харчуванняd | 84,5 % (74,5 %, 91,7 %) | 82,8 % (72,5 %, 90,3 %) |
BSID-III — шкала розвитку немовлят та дітей раннього віку Бейлі — ІІІ видання;
CHOP-INTEND — тест дитячої лікарні Філадельфії для оцінки рухових функцій при нейром’язових захворюваннях у немовлят;
HINE-2 — модуль 2 неврологічного обстеження немовлят за Хаммерсмітом.
a Дані щодо виживаності і виживаності без вентиляції були об’єднані для усіх хворих, які отримали будь-яку дозу рисдипламу в частині 1 і частині 2 дослідження (n = 62). Для етапів моторного розвитку та рухової функції, а також годування, кінцевих точок ефективності дані були об’єднані для усіх хворих, які отримали терапевтичну дозу рисдипламу (усі пацієнти в частині 2 дослідження і пацієнти в когорті отримання високої дози в частині 1; n = 58).
b Визначення відповіді за критерієм HINE-2: відповідь у цьому аналізі визначена як збільшення ≥ 2 бали (або найбільший можливий показник) здатності стукати ніжками АБО збільшення ≥ 1 бал таких етапів розвитку моторної функції, як контроль утримання голови, перевертання, сидіння, повзання, стояння чи хода, ТА поліпшення в більшому числі категорій розвитку моторної функції, ніж погіршення.
c Явище, що відповідає кінцевій точці постійної вентиляції, визначене як трахеостомія або ≥ 16 годин неінвазивної вентиляції легень на день, або інтубація протягом > 21 дня послідовно при відсутності чи після зняття гострого оборотного явища. Чотири пацієнти відповідали критеріям кінцевої точки постійної вентиляції до 24 місяця. Ці чотири пацієнти досягли збільшення такого показника щонайменше на 4 бали за шкалою CHOP-INTEND порівняно з вихідним рівнем.
d Включаючи хворих, які отримували виключно пероральне харчування (41 пацієнт через 12 і 24 місяці), і тих хворих, які отримували пероральне харчування у комбінації із зондом для штучного годування (8 хворих через 12 місяців і 7 хворих через 24 місяці).
Через 24 місяці 40 % (23/58) хворих, які отримували терапевтичну дозу лікарського засобу Еврісді®, могли сидіти без підтримки протягом 30 секунд (BSID-III, пункт 26). Окрім того, пацієнти продовжували досягати додаткові етапи моторного розвитку за показником HINE-2 через 24 місяці; 78 % хворих могли перевертатись (31 % хворих могли перевертатись на бік, 7 % хворих могли перевертатися з положення лежачи на животі в положення лежачи на спині і 40 % хворих могли перевертатися з положення лежачи на спині в положення лежачи на животі) і 28 % хворих змогли досягти сидіння (16 % утримували вагу і 12 % стояли з підтримкою).
Частка живих хворих без потреби постійної вентиляції (виживаність без подій) становила 84 % серед усіх хворих через 24 місяці. Шість немовлят померло (4 протягом перших 3 місяців після включення в дослідження) і у одного пацієнта було передчасно припинене терапія, а через 3,5 місяця після цього пацієнт помер. Чотирьом пацієнтам була необхідна постійна вентиляція до 24 місяця.
СMA із пізнім перебігом
Дослідження BP39055 (SUNFISH) – багатоцентрове дослідження, що містить 2 частин, з вивчення ефективності, безпеки, фармакокінетики та фармакодинаміки лікарський засібу Еврісді® у хворих з діагнозом СMA 2 або 3 типу віком від 2 до 25 років. Частина 1 була дослідницькою частиною з визначення дози і частина 2 – рандомізованою подвійно сліпою плацебо-контрольованою підтверджувальною частиною. Пацієнти з частини 1 не брали участі в частині 2.
Первинна кінцева точка оцінювалась як зміна показника рухової функції (MFM32) до 12 місяця порівняно з вихідним рівнем. MFM32 має змогу оцінити широкий діапазон рухових функцій для широкого кола хворих із СМА. Загальний показник MFM32 виражається у відсотках (діапазон: від 0 до 100) максимального можливого показника, при цьому вищий показник свідчить про більшу рухову функцію. MFM32 визначає здатність до рухової функції, яка стосується важливих щоденних функцій. Незначні зміни рухової функції можуть призвести до значущого поліпшення або втрати щоденної функції(-й).
SUNFISH, частина 2
Частина 2 SUNFISH – це рандомізована подвійно сліпа плацебо-контрольована частина дослідження за участю 180 хворих, які не здатні ходити, зі СМА 2 (71 %) або 3 типу (29 %). Пацієнти були рандомізовані у співвідношенні 2:1 для отримання лікарський засібу Еврісді® в терапевтичній дозі (див. розділ «Спосіб використання та дози») або плацебо. Рандомізація була стратифікована залежно від віку (від 2 до 5 років, від 6 до 11 років, від 12 до 17 років, від 18 до 25 років).
Середній вік хворих на момент початку терапія становив 9 років (діапазон 2–25 років), медіана часу від появи симптомів СМА до першого терапія становила 102,6 (1–275) місяця. Серед учасників дослідження 51 % хворих були жіночої статі, 67 % були європеоїдної раси, 19 % – азіатського походження. На момент початку дослідження 67 % хворих мали сколіоз (32 % з них мали тяжкий сколіоз). Пацієнти мали середній вихідний показник МFM32 46,1 і показник RULM 20,1. Загалом вихідні демографічні характеристики були добре збалансованими між групами прийому лікарський засібу Еврісді® та плацебо, за винятком невідповідності кількості хворих зі сколіозом (63,3 % хворих в групі лікарський засібу Еврісді® та 73,3 % хворих у групі плацебо-контролю).
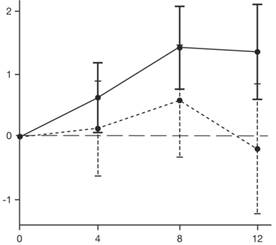
Результати первинного аналізу частини 2 дослідження SUNFISH щодо зміни вихідного загального показника MFM32 через 12 місяців продемонстрували клінічно значущу та статистично достовірну різницю між групами хворих, які отримували терапія лікарський засібом Еврісді® та плацебо. Результати первинного аналізу та основні вторинні кінцеві точки наведено в таблиці 2 та на рисунку 1.
Таблиця 2
Резюме результатів ефективності у хворих із СМА із пізнім початком через 12 місяців терапія (частина 2 дослідження SUNFISH)
| Кінцева точка | Еврісді® (N = 120) | Плацебо (N = 60) |
| Первинна кінцева точка: | ||
| Зміна загального показника MFM321 через 12 місяців порівняно з вихідним рівнем, середньоквадратичне середнє значення (95 %, ДІ) | 1,36 (0,61; 2,11) |
(-1,22; 0,84) |
| Різниця порівняно з плацебо (95 % ДІ), показник p2 | 1,55 (0,30; 2,81) 0,0156 | |
| Вторинні кінцеві точки | ||
| Частка хворих із зміною загального показника MFM321 на 3 або більше через 12 місяців порівняно з вихідним рівнем (95 % ДІ) | 38,3 % (28,9; 47,6) | 23,7 % (12,0; 35,4) |
| Відношення шансів загальної відповіді (95 % ДІ), скориговане (нескориговане) значення p3,4 | 2,35 (1,01; 5,44) 0,0469 (0,0469) | |
| Зміна загального показника RULM5 через 12 місяців порівняно з вихідним рівнем, середньоквадратичне середнє значення (95 % ДІ) | 1,61 (1,00; 2,22) | 0,02 (-0,8; 0,87) |
| Різниця порівняно з плацебо (95 % ДІ), скориговане (нескориговане) значення p2,4 | 1,59 (0,55; 2,62) 0,0469 (0,0028) | |
1 На підставі правила відсутніх даних щодо MFM32 6 хворих були виключені з аналізу (Еврісді®, n = 115; група плацебо-контролю, n = 59).
2 Дані проаналізовано за допомогою змішаної моделі із повторним вимірюванням із вихідним загальним показником, терапіям, візитом, віковою групою, ефектом фактору терапія в залежності від візиту і вихідним в залежності від візиту.
3 Дані аналізувались за допомогою логістичної регресії для вихідного загального показника, групи терапія та вікової групи.
4 Скориговане значення p отримано для кінцевих точок, включених в ієрархічне тестування, і обчислено на підставі усіх значень р для кінцевих точок у порядку ієрархії до поточної кінцевої точки. Нескориговане значення p тестувалось із 5 % рівнем значимості.
5 На підставі правила відсутніх даних щодо RULM 3 пацієнти були виключені з аналізу (Еврісді®, n = 119; група плацебо-контролю, n = 58).
Після завершення 12 місяців терапія 117 хворих продовжили отримувати лікарський засіб Еврісді®. На момент проведення аналізу через 24 місяці пацієнти, які отримували терапія протягом 24 місяців, досягли подальшого поліпшення моторної функції в період між 12 і 24 місяцями терапії. Середня зміна вихідного показника MFM32 порівняно з вихідним рівнем становила 1,83 (ДІ: 0,74–2,92), а RULM – 2,79 (ДІ: 1,94–3,64).
| Зміна середньоквадратичного середнього значення загального показника MFM32 |
|
| Місяці |
 Еврісді®
Еврісді®  Плацебо
Плацебо
* Планка погрішностей означає 95 % довірчий інтервал.
† Загальний показник МFM був обчислений за допомогою керівництва для користувача, виражений як відсоток максимального можливого показника для шкали (тобто суму балів для 32 пунктів ділили на 96 та множили на 100).
Рис. 1. Середня зміна (LS) загального показника MFM32 від вихідного рівня через 12 місяців в частині 2 дослідження SUNFISH.
SUNFISH, частина 1
Ефективність лікарський засібу Еврісді® у хворих із СМА із пізнім початком також підтверджується результатами частини 1 дослідження SUNFISH з визначення дози. У частину 1 було включено 51 пацієнта із СMA 2 та 3 типу (зокрема 7 хворих, які здатні ходити) віком 2–25 років. Через один рік терапія терапевтичною дозою (дозою, обраною для частини 2) спостерігалось клінічно значиме поліпшення моторної функції за результатами MFM32 із середньою зміною від вихідного рівня 2,7 бала (95 % ДІ: 1,5; 3,8). Поліпшення MFM32 підтримувалось протягом періоду до 2 років терапія лікарський засібом Еврісді® (середня зміна 2,7 бала (95 % ДІ: 1,2; 4,2)).
У пошуковому аналізі рухова функція, оцінена за допомогою MFM, порівнювалась у частині 1 SUNFISH та історичному контролі із природним прогресуванням захворювання (зваженим на основі основних прогностичних факторів). Зміна загального показника MFM порівняно з вихідним рівнем через 1 і 2 роки була більшою у хворих, які отримували Еврісді®, порівняно з когортою природного перебігу (через 1 рік: різниця в 2,7 бала; p < 0,0001; через два роки: різниця в 4 бали; p < 0,0001). У когорті природного перебігу спостерігалось зниження моторної функції, що очікується з огляду на природне прогресування СMA (середня зміна через 1 рік: –0,6 бала; через 2 роки: –2 бали).
Пресимптоматична СМА
Дослідження BN40703 (RAINBOWFISH) — одногрупове відкрите багатоцентрове дослідження, що триває, з вивчення ефективності, безпеки, фармакокінетики та фармакодинаміки лікарського засобу Еврісді® у немовлят віком від народження до 6 тижнів (на момент отримання першої дози), яким встановлено генетичний діагноз СMA, однак прояви ще відсутні.
На момент проміжного аналізу загалом 18 хворих із СМА до виникнення симптомів було включено в дослідження RAINBOWFISH. Попередньо ефективність у хворих із СМА до виникнення симптомів вивчалась у 7 хворих, які отримували терапія лікарський засібом Еврісді® протягом щонайменше 12 місяців. Середній вік хворих на момент отримання першої дози становив 35 днів (16–40 днів), 71 % були жіночої статі, 100 % — європеоїдної раси. 4 пацієнти мали 2 копії гена SMN2, 2 пацієнта — 3 копії гена SMN2 і 1 пацієнт — 4 або більше копії гена SMN2.
На момент проведення проміжного аналізу пацієнти із 2 або 3 копіями SMN2 (N = 6) досягли таких результатів за показником HINE-2 через 12 місяців: 6 (100 %) хворих могли сидіти (5 хворих могли самостійно перевертатися і 1 пацієнт сидів стабільно), 4 (67 %) пацієнти могли стояти (3 пацієнти могли стояти без сторонньої допомоги і 1 пацієнт міг стояти з допомогою) і 3 (50 %) пацієнти могли ходити самостійно.
Використання пацієнтам, які раніше отримували терапія іншими засобами, що модифікують перебіг СМА
Дослідження BP39054 (JEWELFISH) – одногрупове відкрите дослідження безпеки, переносимості, фармакокінетики та фармакодинаміки лікарський засібу Еврісді® за участю хворих із інфантильною формою СМА або формою СМА з пізнім початком віком від 6 місяців до 60 років, які раніше отримували терапія з приводу СМА (у тому числі нусинерсен та онасемноген абепарвовек). Зі 173 хворих, які отримали лікарський засіб Еврісді®, 76 хворих отримували попереднє терапія нусинерсеном (9 хворих із СMA 1 типу, 43 пацієнти із СМА 2 типу і 24 пацієнти із СMA 3 типу) і 14 хворих раніше отримували терапія онасемногеном абепарвовеком (4 пацієнти із СМА 1 типу і 10 хворих із СМА 2 типу). Середній вік хворих на початку терапія лікарський засібом Еврісді® становив 14 років (діапазон 1–60 років).
На момент включення в дослідження зі 168 хворих віком 2–60 років 83 % мали сколіоз (39 % – тяжкий сколіоз) і у 63 % показник за розширеною шкалою оцінки рухової функції за Хаммерсмітом (HFMSE) становив < 10 балів. В дослідження також були включені 15 хворих, здатних пересуватися самостійно (віком 5–46 років).
Пошукові результати ефективності оцінювались шляхом вимірювання рухової функції відповідно до віку, у тому числі за допомогою шкал MFM-32 та RULM для хворих віком 2–60 років, BSID-III та HINE-2 для хворих віком до 2 років та тесту шестихвилинної ходи (6MWT) для хворих віком ≥ 6 років, здатних пересуватися самостійно. За результатами первинного аналізу після 24 місяців терапія у хворих віком 2–60 років спостерігалась загальна стабілізація рухової функції за шкалами MFM-32 та RULM (n = 137 і n = 133 відповідно). Пацієнти віком до 2 років (n = 6) зберегли або покращили розвиток рухової функції, зокрема утримання голови, перевертання, сидіння без підтримки. Результати 6MWT демонструють середнє поліпшення на 30,88 метра (95 % ДІ: -5,54, 67,29, n = 8). Усі пацієнти, здатні пересуватись самостійно, зберегли здатність ходити.
Фармакокінетика.
Фармакокінетичні параметри рисдипламу були охарактеризовані у здорових дорослих осіб та у хворих із СMA.
Після прийому орального розчину лікарський засібу Еврісді® у дозах від 0,6 до 18 мг фармакокінетика рисдипламу була приблизно лінійною. Фармакокінетика рисдипламу була найкраще описана за допомогою популяційної ФК моделі зі всмоктуванням з трикамерним транзитом, двокамерним розподілом та виведенням першого порядку. Було виявлено, що маса тіла та вік пацієнта мають суттєвий вплив на фармакокінетику лікарський засібу.
Розрахункова експозиція (середня AUC0–24h) у хворих з інфантильною формою СМА (віком 2–7 місяців на момент включення в дослідження) при рекомендованій дозі 0,2 мг/кг один раз на добу становила 1930 нг.год/мл. Середня розрахункова експозиція у немовлят віком від 16 днів до < 2 місяців із пресимптоматичною СМА в дослідженні RAINBOWFISH становила 2080 нг.год/мл після 2 тижнів щоденного прийому в дозі 0,15 мг/кг.
Розрахункова експозиція у хворих з формою СМА з пізнім початком (віком 2–25 років на момент включення в дослідження) в дослідженні SUNFISH (частина 2) при терапевтичній дозі (0,25 мг/кг один раз на добу пацієнтам з масою тіла < 20 кг; 5 мг один раз в день пацієнтам із масою тіла ≥ 20 кг) становила 2010 нг.год/мл. Максимальна концентрація, що спостерігалася (середня Cmax), становила 194 нг/мл при дозуванні 0,2 мг/кг в дослідженні FIREFISH і 120 нг/мл в частині 2 дослідження SUNFISH. Середня розрахункова максимальна концентрація при дозуванні 0,15 мг/кг в дослідженні RAINBOWFISH становить 113 нг/мл.
Всмоктування
Рисдиплам швидко всмоктувався при пероральному прийомі натще, при цьому tmax в плазмі крові варіював від 1 до 4 годин. У клінічних дослідженнях рисдиплам застосовували вранці з прийомом їжі або після лактації.
Розподіл
Розраховані популяційні фармакокінетичні показники становили: 98 л – очевидний центральний об’єм розподілу, 93 л – периферичний об’єм і 0,68 л/годину – міжкомпартментний кліренс.
Рисдиплам в основному зв’язується з альбумінами сироватки крові, не зв’язуючись з альфа-1 кислими глікопротеїнами, при цьому вільна фракція становить 11 %.
Mетаболізм
Рисдиплам в основному метаболізується за допомогою флавін-монооксигенази 1 та 3 (FMO1 і FMO3), a також ізоферментами CYP 1A1, 2J2, 3A4 і 3A7. Вихідний лікарський засіб був основним компонентом, виявленим у плазмі, і становив у кровообігу 83 % речовини, пов’язаної з лікарським засобом. Фармакологічно неактивний метаболіт M1 був ідентифікований як основний циркулюючий метаболіт.
Виведення
Популяційний фармакокінетичний показник очевидного кліренсу (CL/F) рисдипламу становив 2,6 л/год. Ефективний період напіввиведення рисдипламу становив близько 50 годин у хворих із СМА.
Приблизно 53 % дози (14 % у формі незміненого рисдипламу) виводилось з калом та 28 % із сечею (8 % у формі незміненого рисдипламу).
Особливі групи хворих
Розлади функції печінки
Розлади функції печінки легкого та помірного ступеня не впливало на ФK рисдипламу. Після прийому 5 мг рисдипламу середні співвідношення Cmax та AUC становили 0,95 і 0,80 у осіб із легким (n = 8) та 1,20 і 1,08 у хворих з помірним розладим функції печінки (n = 8) порівняно із відповідними показниками у хворих з нормальною функцією печінки (контрольна група, n = 10). Безпека і ФК у хворих із тяжким розладим функції печінки на сьогодні не вивчались.
Розлади функції нирок
Дослідження ФК рисдипламу у хворих із розладим функції нирок не проводились. Виведення рисдипламу у вигляді незміненої речовини нирками є незначним (8 %).
Пацієнти літнього віку
Спеціальні дослідження фармакокінетики лікарський засібу Еврісді® у хворих із СМА віком понад 60 років не проводились. Пацієнти із СМА віком до 60 років були включені в дослідження JEWELFISH. Пацієнти без СМА віком до 69 років були включені в клінічні дослідження ФК.
Діти
Маса тіла та вік пацієнта були ідентифіковані як коваріати в популяційному ФК аналізі. Тому дозу коригують залежно від віку (менше та більше 2 місяців та 2 років) та маси тіла (до 20 кг) з метою отримання подібної експозиції в різних вікових групах та вагових категоріях. Стосовно хворих віком до 16 днів дані відсутні.
Етнічна приналежність
Фармакокінетика рисдипламу не відрізняється у японців та хворих європеоїдної раси.
Терапія 5q-асоційованої спінальної м’язової атрофії (СMA) у дітей і дорослих хворих.
Відома гіперчутливість до рисдипламу або будь-якої з допоміжних речовин, зазначених у розділі «Склад».
Вплив Еврісді® на інші медикаменти
In vitro рисдиплам та його основний циркулюючий метаболіт M1 не індукували CYP1A2, 2B6, 2C8, 2C9, 2C19 та 3A4. In vitro рисдиплам та M1 не пригнічували (оборотне або залежне від часу пригнічення) будь-який з ферментів CYP, що досліджувалися (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6), за винятком CYP3A.
Лікарський засіб Еврісді® є слабким інгібітором CYP3A. У здорових дорослих осіб прийом лікарський засібу Еврісді® один раз на добу протягом 2 тижнів дещо підвищував експозицію мідазоламу, високочутливого субстрату CYP3A (AUC 11 %; Cmax 16 %). Така взаємодія не вважається клінічно значимою, тому корекція дози субстратів CYP3A не потрібна.
З огляду на результати використання фармакокінетичної моделі на основі фізіології (PBPK), подібний ефект очікується у дітей та немовлят віком від 2 місяців.
Дослідження іn vitro продемонстрували, що рисдиплам та його основний метаболіт не є суттєвими інгібіторами MDR1 людини, поліпептиду-транспортера органічних аніонів (OATP)1B1, OATP1B3, транспортера органічних аніонів 1 та 3 (OAT 1 та 3). Однак рисдиплам та його метаболіт in vitro є інгібіторами поліпептиду-транспортера органічних катіонів 2 (OCT2) людини і транспортерів білків множинної резистентності та виведення токсинів (MATE)1 та транспортерів MATE2-K. За умови використання в терапевтичних концентраціях взаємодія лікарського засобу з субстратами OCT2 не очікується. Вплив супутнього використання рисдипламу на фармакокінетику субстратів MATE1 та MATE2-K у людини невідомий. На підставі даних in vitro, лікарський засіб Еврісді® може збільшувати плазмову концентрацію медикаментів, які виводяться за допомогою MATE1 або MATE2-K, наприклад метформіну (див. розділ «Фармакокінетика»). Якщо неможливо уникнути супутнього використання, слід здійснювати моніторинг щодо пов’язаної з лікарським засобом токсичності і розглянути питання про зниження дози іншого одночасно застосовуваного лікарський засібу за умови потрібності.
Вплив інших медикаментів на Еврісді®
Рисдиплам метаболізується в основному за участю флавін-монооксигенази 1 та 3 (FMO1 та 3), а також за допомогою CYPs 1A1, 2J2, 3A4 і 3A7. Рисдиплам не є субстратом білка множинної резистентності 1 людини (MDR1).
При одночасному застосуванні сильного інгібітору CYP3A ітраконазолу у дозі 200 мг двічі на добу та рисдипламу у дозі 6 мг одноразово перорально не спостерігалося клінічно значущого впливу на фармакокінетику (ФK) рисдипламу (збільшення AUC на 11 %, зниження Cmax на 9 %). При одночасному застосуванні з інгібітором CYP3A корекція дози лікарський засібу Еврісді® не потрібна.
Не очікується взаємодії з іншими лікарськими засобами через FMO1- та FMO3-опосередковані шляхи передачі сигналу.
Загальні
У дослідженнях на тваринах спостерігались зміни з боку сітківки, епітелію, особливо з боку шкіри та шлунково-кишкового тракту, та прояви токсичності з боку кісткового мозку (зміни в клінічному аналізі крові). На сьогодні ризик таких змін у людей не можна остаточно оцінити через обмежені довгострокові дані про безпеку.
Ембріофетальна токсичність
У дослідженнях на тваринах спостерігалася ембріофетальна токсичність. Хворих резасібивного віку слід проінформувати про ризики. Потрібно використовувати високоефективні методи контрацепції у період терапія та щонайменше протягом 1 місяця після прийому останньої дози лікарський засібу Еврісді® жінками та протягом 4 місяців після прийому останньої дози лікарський засібу Еврісді® чоловіками (див. розділ «Спосіб використання та дози»).
Потенційний вплив на фертильність чоловіків
З огляду на оборотні ефекти лікарський засібу Еврісді® на чоловічу фертильність, що спостерігалися у дослідженнях на тваринах, пацієнтам-чоловікам не слід бути донорами сперми у період терапія та протягом 4 місяців після прийому останньої дози лікарський засібу Еврісді® (див. розділ «Фармакокінетика»).
Слід уникати контакту порошку та відновленого орального розчину зі шкірою. При потраплянні лікарського засобу (порошку або розчину) на шкіру уражену ділянку потрібно промити водою з милом.
Додаткові компоненти.
Цей лікарський засіб має у складі 0,38 мг натрію бензоату в 1 мл. Цей лікарський засіб має у складі менше 1 ммоль натрію (23 мг) на дозу, тобто вважається вільним від натрію.
Цей лікарський засіб має у складі ізомальт. Пацієнтам із рідкісною вродженою непереносимістю фруктози не слід використовувати цей лікарський засіб.
Використання в час вагітності або лактації.
З огляду на результати доклінічних досліджень, фертильність чоловіків може порушуватися у період терапія лікарський засібом Еврісді®. У резасібивних органах щурів та мавп спостерігалися дегенерація сперми та зниження кількості сперматозоїдів.
З пацієнтами чоловічої статі перед початком терапія лікарський засібом Еврісді® потрібно обговорити стратегії збереження фертильності. Пацієнти чоловічої статі можуть розглянути ймовірність збереження сперми до початку терапія або після періоду без терапія протягом не менше 4 місяців (див. розділ «Особливості використання»).
З огляду на результати доклінічних досліджень, впливу лікарський засібу Еврісді® на фертильність жінок не очікується.
Жінок резасібивного віку слід обстежити щодо вагітності до початку терапія лікарський засібом Еврісді®.
Пацієнтам чоловічої та жіночої статі резасібивного віку слід дотримуватися таких вимог щодо контрацепції:
Вагітність
Відсутні клінічні дані щодо використання лікарський засібу Еврісді® вагітним. Рисдиплам продемонстрував ембріофетотоксичну та тератогенну дію у тварин. З огляду на дані, отримані в дослідженнях на тваринах, рисдиплам проникає через плацентарний бар’єр і може спричиняти ураження плода.
Лікарський засіб Еврісді® не слід використовувати у час вагітності, якщо тільки у цьому немає чіткої потрібності. Якщо вагітна жінка потребує терапія лікарський засібом Еврісді®, їй потрібно чітко пояснити потенційний ризик для плода.
Годування груддю
Невідомо, чи екскретується лікарський засіб Еврісді® в грудне молоко людини. Дослідження на щурах показали, що рисдиплам екскретується в грудне молоко. Оскільки потенціал нанесення шкоди немовляті, який знаходиться на грудному вигодовуванні, невідомий, лікуючий лікар повинен прийняти рішення щодо подальшої терапії пацієнта. У період терапія лікарський засібом Еврісді® лактації не рекомендується.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Вплив лікарський засібу Еврісді® на швидкість реакції при керуванні автотранспортом або іншими механізмами не вивчався у відповідних дослідженнях.
Оральний розчин лікарський засібу Еврісді® повинен готувати медичний фахівець (тобто лікар або фармацевт) перед відпуском пацієнту.
Медичному працівнику потрібно перед використанням першої дози обговорити з пацієнтом або особою, яка доглядає за пацієнтом, як приготувати та прийняти призначену добову дозу (див. нижче «Інструкції щодо поводження»).
Починати та контролювати терапія лікарський засібом Еврісді® повинні лікарі, які мають досвід діагностики та терапія хворих зі спінальною м’язовою атрофією.
Програма клінічної розробки не включає хворих із СМА IV типу.
Рекомендоване дозування
Лікарський засіб Еврісді® приймають перорально один раз на добу, приблизно в один і той же час щодня, використовуючи шприци для орального прийому для повторного використання, що надаються в упаковці. Рекомендована доза лікарський засібу Еврісді® для терапія СМА визначається залежно від віку та маси тіла пацієнта (див. таблицю 3).
Tаблиця 3
Режим дозування залежно від віку та маси тіла
| Віка та маса тіла | Рекомендована добова доза |
| від 16 днів до < 2 місяців | 0,15 мг/кг |
| від 2 місяців до < 2 років | 0,20 мг/кг |
| ≥ 2 років та маса тіла < 20 кг | 0,25 мг/кг |
| ≥ 2 років та маса тіла ≥ 20 кг | 5 мг |
а На підставі скоригованого віку для недоношених немовлят.
Зміну дози потрібно здійснювати під наглядом медичного працівника. Терапія добовою дозою вище 5 мг дотепер не вивчалось. Немає даних щодо використання немовлятам віком до 16 днів.
Пацієнти з розладим функції печінки
Пацієнтам із легким чи помірним розладим функції печінки корекція дози не потрібна. Не вивчалося використання лікарський засібу Еврісді® пацієнтам із тяжким розладим функції печінки (див. розділ «Фармакокінетика»).
Пацієнти з розладим функції нирок
Безпека та ефективність використання лікарський засібу Еврісді® пацієнтам із розладим функції нирок не вивчались. Не очікується потрібності корекції дози пацієнтам із розладим функції нирок (див. розділ «Фармакокінетика»).
Пацієнти літнього віку
Клінічні дослідження лікарський засібу Еврісді® не включали хворих віком від 65 років, тому не було встановлено різниці між їхньою відповіддю на терапія та відповіддю молодших хворих.
Діти
Наявні обмежені дані щодо безпеки та ефективності лікарського засобу Еврісді® у дітей віком до 2 місяців. Немає даних щодо недоношених немовлят або новонароджених віком до 16 днів (див. підрозділ «Клінічна ефективність» вище).
Відкладений прийом
Лікарський засіб Еврісді® приймають перорально один раз на добу приблизно в один і той же час щодня. Якщо прийом дози лікарський засібу Еврісді® пропущено, лікарський засіб Еврісді® слід прийняти якомога швидше, якщо затримка становить не більше 6 годин від запланованого прийому, і звичайний режим дозування можна відновити наступного дня. В іншомза умови не слід вживати пропущену дозу та прийняти наступну дозу в запланований час наступного дня.
Якщо доза не проковтнулася повністю або виникло блювання після прийому лікарський засібу Еврісді®, не слід вживати іншу дозу, щоб компенсувати втрачену дозу. Слід зачекати наступного дня для прийому наступної дози в запланований час.
Спосіб використання
Для прийому добової дози Еврісді® потрібно використовувати оральний шприц для багаторазового використання, що надається в картонній упаковці разом із лікарським засобом (див. таблицю 4).
Таблиця 4
Вибір відповідного орального шприца для багаторазового використання для прийому призначеної добової дози Еврісді®
| Розмір шприца | Об’єм дозування | Ціна поділки шприца |
| 1 мл | від 0,3 до 1 мл | 0,01 мл |
| 6 мл | від 1 до 6 мл | 0,1 мл |
| 12 мл | від 6,2 до 6,6 мл | 0,2 мл |
Для розрахунку об’єму дозування також потрібно врахувати ціну поділки шприца. Округляйте об’єм дози в бік збільшення або зниження до найближчої ціни поділки, позначеної на вибраному оральному шприці (наприклад, з 6,3 мл до 6,4 мл, з 3,03 мл до 3 мл і з 1,05 до 1,1 мл).
Лікарський засіб Еврісді® у вигляді розчину потрібно прийняти відразу після його набору в оральний шприц для багаторазового використання. Якщо вміст шприца не прийнятий протягом 5 хвилин, слід звільнити оральний шприц від лікарський засібу (див. нижче «Утилізація невикористаного лікарського засобу/лікарського засобу, строк придатності якого минув») і приготувати нову дозу.
Лікарський засіб Еврісді® потрібно вживати після прийому їжі. Пацієнту потрібно випити води після прийому Еврісді®, щоб переконатися, що лікарський засіб повністю проковтнувся. Якщо пацієнт не може ковтати і йому встановлено назогастральний зонд або гастростомічну трубку, Еврісді® можна вводити через зонд/трубку. Після введення лікарський засібу зонд/трубку слід промити водою (див. нижче «Інструкції щодо поводження»).
Інструкції щодо поводження
Інструкції, яких потрібно дотримуватися до, у період та після приготування орального розчину:
Пацієнт або особа, що доглядає за пацієнтом, повинні бути проінструктовані медичним працівником, як слід готувати та вживати призначену добову дозу перед доставкою приготовленого розчину.
Приготування орального розчину
Наливають 79 мл очищеної води або води для ін’єкцій в пляшку із лікарським засобом.
Вставляють втискний адаптер для пляшки в отвір пляшки, протискуючи його вниз.
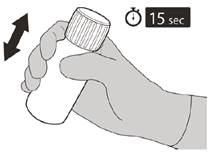
Після повного закриття пляшки струшуйте протягом 15 секунд.
Через 10 хвилин повинен бути отриманий прозорий розчин. Якщо розчин не став прозорим, його слід струсити ще протягом 15 секунд.
Потрібно вирахувати 64 дні після приготування розчину. День приготування розчину вважається днем 0. Розраховану дату слід зазначити на етикетці пляшки та картонній коробці у полі: Відновлений оральний розчин ВИКИНУТИ ПІСЛЯ: (день/місяць/рік).
Утилізація невикористаного лікарського засобу/лікарського засобу, строк придатності якого минув
Потрапляння медикаментів в навколишнє середовище має бути зведено до мінімуму. Не можна утилізувати медикаменти через стічні води, а також слід уникати утилізації з побутовими відходами.
Невикористаний лікарський засіб/лікарський засіб із терміном дії, що закінчився, повинен бути утилізований професійно в місці видачі лікарського засобу (лікарем або фармацевтом).
Інструкція з приготування розчину (ЛИШЕ ДЛЯ МЕДИЧНИХ ПРАЦІВНИКІВ, ЗОКРЕМА ЛІКАРІВ АБО ФАРМАЦЕВТІВ)
Оральний розчин Еврісді® повинен бути приготований медичним працівником перед відпуском пацієнту.
|
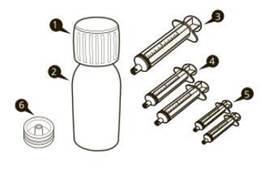
Одна упаковка лікарський засібу Еврісді® має у складі (див. рисунок A): 1) 1 кришка 2) 1 пляшка з лікарський засібом Еврісді® 3) 1 оральний шприц для багаторазового використання по 12 мл (у пакетику) 4) 2 оральні шприци для багаторазового використання по 6 мл (у пакетиках) 5) 2 оральні шприци для багаторазового використання по 1 мл (у пакетиках) 6) 1 втискний адаптер для пляшки 7) 1 інструкція для медичного використання лікарського засобу (не показана) |
Рисунок A |
Важлива інформація про лікарський засіб Еврісді®
Як зберігати лікарський засіб Еврісді®
Приготування розчину
|
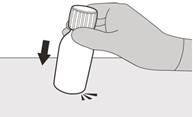
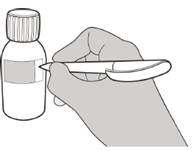
Рисунок В | Крок 1 Обережно постукати по дну пляшки, щоб розпушити порошок (див. рисунок B). |
|
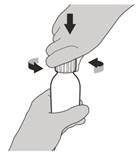
Рисунок С | Крок 2 Зняти кришку, натиснувши на неї вниз, а потім скручуючи вліво (проти годинникової стрілки) (див. рисунок C). Не викидати кришку. |
|
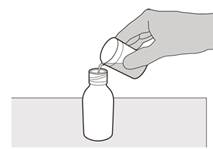
Рисунок D | Крок 3 Обережно додати 79 мл води очищеної чи води для ін’єкцій у пляшку з лікарський засібом (див. рисунок D). |
|
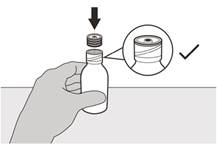
Рисунок Е | Крок 4 Тримати пляшку з лікарський засібом однією рукою на столі. Вставити втискний адаптер для пляшки в отвір пляшки, натискаючи на нього донизу іншою рукою доки адаптер не буде повністю притиснутий до краю отвору пляшки (див. рисунок E). |
|
Рисунок F | Крок 5 Надіти кришку назад на пляшку. Повернути кришку вправо (за годинниковою стрілкою), щоб закрити пляшку. Переконатись, що пляшка повністю закрита, а потім добре струшувати протягом 15 секунд (див. рисунок F). Зачекати 10 хвилин. Ви повинні отримати прозорий розчин. Якщо ні, ще раз добре струшувати протягом 15 секунд. |
|
Рисунок G | Крок 6 Визначити дату «ВИКИНУТИ ПІСЛЯ: (день/місяць/рік)», вирахувавши 64 дні після приготування розчину (примітка: день приготування обчислюється як день 0. Наприклад, якщо Ви приготували розчин 1 квітня, датою «ВИКИНУТИ ПІСЛЯ» буде 4 червня). Зазначити цю дату у відповідному полі на етикетці пляшки (див. рисунок G) та картонній коробці: Відновлений оральний розчин ВИКИНУТИ ПІСЛЯ: (день/місяць/рік). Помістити пляшку назад у картонну коробку разом зі шприцами (у пакетиках) та інструкцією для медичного використання. Все зберігати в холодильнику, тримаючи пляшку у вертикальному положенні. |
Відсутній досвід передозування лікарський засібом Еврісді® в клінічних дослідженнях. Відсутній відомий антидот при передозуванні лікарський засібом Еврісді®. При передозуванні слід уважно спостерігати за станом пацієнта та розпочати підтримувальну терапію.
Класифікація частоти виникнення побічних реакцій лікарського засобу: дуже часто (≥ 1/10), часто (від ≥ 1/100 до < 1/10), нечасто (від ≥ 1/1 000 до < 1/100), рідко (від ≥ 1/10 000 до < 1/1000), дуже рідко (< 1/10 000); частота невідома (не можна розрахувати на основі наявних даних).
Коротка характеристика профілю безпеки
У хворих із СМА з інфантильним початком найбільш поширеними побічними реакціями, що спостерігались в клінічних дослідженнях лікарського засобу Еврісді®, були підвищення температури тіла (54,8 %), висипання (29 %) та діарея (19,4 %). У хворих із СМА із більш пізнім початком найбільш поширеними побічними реакціями, що спостерігались в клінічних дослідженнях лікарського засобу Еврісді®, були підвищення температури тіла (21,7 %), головний біль (20 %), діарея (16,7 %) і висипання (16,7 %).
Зазначені вище небажані реакції виникали без ідентифікованих клінічних особливостей або характеру зв’язку із часом і загалом зникали без припинення терапія у хворих із СМА з інфантильним початком та пізнім початком.
Таблиця 6
Резюме побічних реакцій у хворих із СМА з інфантильним початком та пізнім початком, що спостерігались в клінічних дослідженнях лікарський засібу Еврісді®
| Клас системи органів | СMA з інфантильним початком2 (тип 1) | СMA із пізнім початком3 (тип 2 та 3) |
| Шлунково-кишкові розлади | ||
| Діарея | Дуже часто | Дуже часто |
| Нудота | Не застосовне | Часто |
| Виразки на слизовій оболонці ротової порожнини та афтозні виразки | Часто | Часто |
| Розлади з боку шкіри та підшкірної клітковини | ||
| Висипання1 | Дуже часто | Дуже часто |
| Розлади з боку нервової системи | ||
| Головний біль | Не застосовне | Дуже часто |
| Загальні розлади та реакції в місці введення | ||
| Підвищення температури тіла (включаючи гіперпірексію) | Дуже часто | Дуже часто |
| Інфекції та інвазії | ||
| Інфекція сечовивідних шляхів (включаючи цистит) | Часто | Часто |
| Розлади з боку кістково-м’язової системи та сполучної тканини | ||
| Артралгія | Не застосовно | Часто |
1 Включає дерматит, акнеформний дерматит, алергічний дерматит, еритему, фолікуліт, висипання, еритематозне висипання, макулопапульозне висипання, папульозне висипання.
2 У хворих із СМА з інфантильним початком (FIREFISH, частини 1 та 2) небажані реакції визначено як явища, які відбулись у 2 % хворих або більше і причинний зв’язок яких із лікарський засібом Еврісді® є можливим.
3 У хворих із СМА із пізнім початком (SUNFISH, частина 2) небажані реакції визначено як явища, які відбулись щонайменше на 2 % частіше у хворих, які отримували терапія лікарський засібом Еврісді®, порівняно з такими у групі плацебо у період подвійно контрольованого сліпого періоду і причинний зв’язок яких із лікарський засібом Еврісді® є можливим.
Наявні дані з безпеки обмежені стосовно числа хворих, які отримували лікарський засіб Еврісді®, та тривалості експозиції. Можуть спостерігатись потенційні відносно рідкісні та потенційно серйозні небажані реакції (ПР), які не були виявлені у період програми дослідження.
В дослідженні RAINBOWFISH отримані обмежені дані щодо безпеки використання лікарського засобу Еврісді® для новонароджених та немовлят із пресимптоматичною СМА. Дослідження RAINBOWFISH — відкрите одногрупове дослідження, що триває. У період проведення проміжного аналізу в дослідження було включено 18 хворих віком 16 – 40 днів на момент отримання першої дози лікарського засобу Еврісді®. Маса тіла хворих становила від 3,1 до 5,7 кг. Середня тривалість експозиції становила 8,7 місяця (діапазон від 0,5 до 22,8 місяця) (див. підрозділ «Клінічна ефективність» вище). Відповідно до проміжних даних з безпеки профіль безпеки лікарського засобу Еврісді® у хворих із СМА до виникнення симптомів є порівнянним із таким у хворих із симптоматичною СМА з інфантильним початком та хворих із СМА з пізнім початкам. Довгострокові дані на цей час відсутні.
Профіль безпеки у хворих, які раніше отримували терапія іншими засобами, що модифікують перебіг СМА
На підставі первинного аналізу учасників дослідження JEWELFISH профіль безпеки лікарський засібу Еврісді® у раніше лікованих хворих, які отримували терапія лікарський засібом Еврісді® протягом періоду до 59 місяців у дослідженні JEWELFISH, узгоджується із профілем безпеки при лікуванні раніше не лікованих хворих із СМА, які отримували лікарський засіб Еврісді® в дослідженнях FIREFISH (частини 1 і 2), SUNSH (частини 1 і 2) та RAINBOWFISH.
Пацієнти, які раніше отримували нусинерсен (n = 76) або онасемноген абепарвовек (n = 14), були включені в дослідження JEWELFISH (див. підрозділ «Клінічна ефективність»).
Доклінічні ефекти
Доклінічні ефекти стосовно структури сітківки, епітеліальної тканини та гематологічних параметрів, описані у розділі «Доклінічні дані», на сьогодні не спостерігались в клінічних дослідженнях лікарський засібу Еврісді® при СМА.
Подовження QT
Аналіз фармакокінетики/фармакодинаміки показав відсутність ознак подовження QTc в результаті використання лікарський засібу Еврісді® із експозицією в терапевтичному діапазоні, однак немає відповідних даних при застосуванні лікарський засібу Еврісді® при експозиції, що перевищує терапевтичні рівні.
Досвід післяреєстраційного використання
У період післяреєстраційного використання спостерігався шкірний васкуліт, прояви якого зникли після остаточної відміни лікарського засобу Еврісді®. На основі наявних даних неможливо визначити категорію захворюваності і частоту виникнення цієї побічної реакції.
2 роки.
Після відновлення готовий до використання оральний розчин стабільний протягом 64 днів при зберіганні в холодильнику при температурі 2–8 ºС. За потрібності пацієнт або особа, яка за ним доглядає, можуть зберігати оральний розчин при кімнатній температурі (нижче 40 °C) сумарно не більше ніж 5 днів.
Зберігати порошок для орального розчину при температурі не вище 25 ºС у заводській упаковці з метою захисту від світла та вологи. Зберігати у недоступному для дітей місці.
Зберігати готовий до використання оральний розчин в холодильнику при температурі 2–8 ºС у заводській упаковці з метою захисту від світла. Тримати пляшку щільно закритою та завжди зберігати у вертикальному положенні.
Не спостерігалось несумісності між лікарський засібом Еврісді® та рекомендованими шприцами для багаторазового використання для орального введення лікарський засібу.
Скляна пляшка об’ємом 100 мл бурштинового кольору (клас ІІІ відповідно до Фарм. США та Євр. Фарм.) з кришкою білого кольору із функцією захисту від розкриття дітьми (зовнішня оболонка із поліетилену високої щільності; внутрішня оболонка із гомополімеру поліпропілену) з кільцем контролю першого розкриття та вкладкою з поліетилену та полівініліденхлориду у комплекті з 1 втискним адаптером для пляшки, 2 оральними шприцами для багаторазового використання об’ємом 1 мл (кожний у поліетиленовому пакетику), 2 оральними шприцами для багаторазового використання об’ємом 6 мл (кожний у поліетиленовому пакетику) та 1 оральним шприцом для багаторазового використання об’ємом 12 мл (у поліетиленовому пакетику), які вміщені у поліетиленовий пакет. По 1 пляшці та 1 комплекту у картонній коробці.
За рецептом.
Ф.Хоффманн-Ля Рош Лтд
Місцезнаходження виробника та адреса місця провадження його діяльності.
Вурмісвег, 4303 Кайсераугст, Швейцарія

Поки що відгуків немає
Будьте першими, хто розповість про свій досвід