Нам важлива ваша думка!
Відгук кожного пацієнта — це вклад у формування чесного рейтингу Довіри про медицину України

діюча речовина: pralsetinib;
1 капсула містить пралсетинібу 100 мг;
допоміжні речовини: гідроксипропілметилцелюлоза, целюлоза мікрокристалічна, натрію гідрокарбонат, кислота лимонна безводна, магнію стеарат, крохмаль прежелатинізований;
оболонка капсули: гіпромелоза, титану діоксид (Е 171), FD&C синій № 1 (Е 133), чорнило для друку.
Капсули.
Основні фізико-хімічні властивості: капсула розміром 0 з непрозорою оболонкою світло-блакитного кольору з білим відбитком «BLU-667» на корпусі та білим відбитком «100 mg» на кришечці, що містить порошок від білого до майже білого кольору.
Антинеопластичні засоби. Інгібітори протеїнкінази.
Код АТХ L01E X23.
Фармакодинаміка.
Механізм дії
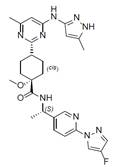
Пралсетиніб є інгібітором рецептора тирозинкінази. Хімічна назва пралсетинібу — (cis)-N-((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-(5- methyl-1H-pyrazol-3-ylamino)pyrimidin-2-yl)cyclohexanecarboxamide. Молекулярна формула пралсетинібу — C27H32FN9O2. Молекулярна маса становить 533,61 г/моль. Пралсетиніб має таку структуру:
Розчинність пралсетинібу у водному середовищі зменшується в діапазоні pH від 1,99 до 7,64 з 0,880 мг/мл до < 0,001 мг/мл, що свідчить про зменшення розчинності зі зростанням pH.
Пралсетиніб є інгібітором кіназ дикого типу RET і онкогенного злиття RET (CCDC6-RET), а також мутацій (RET V804L, RET V804M і RET M918T) із напівмаксимальною інгібуючою концентрацією (IC50s) менше 0,5 нМ. В очищеному ферментному аналізі пралсетиніб інгібував DDR1, TRKC, FLT3, JAK1-2, TRKA, VEGFR2, PDGFRB і FGFR1 в вищих концентраціях, які все ще були клінічно досяжними при Cmax. В оцінці стану клітин пралсетиніб інгібував RET в концентраціях, приблизно в 14, 40 і 12 разів менших, ніж VEGFR2, FGFR2 і JAK2 відповідно.
Певні білки злиття RET і активуючі точкові мутації можуть стимулювати онкогенний вплив через гіперактивацію низхідних сигнальних шляхів, що призводить до неконтрольованої проліферації клітин. Пралсетиніб продемонстрував протипухлинну активність в культивованих клітинах і тваринних моделях імплантації пухлини, що експресують онкогенні злиття RET або мутації, які включають KIF5B-RET, CCDC6-RET, RET M918T, RET C634W, RET V804E, RET V804L і RET V804M. Окрім того, пралсетиніб подовжував виживаність у мишей із внутрішньочерепними імплантатами моделей пухлин, що експресують KIF5B-RET або CCDC6-RET.
Фармакодинаміка
Взаємозв’язок експозиція — відповідь і часова динаміка фармакодинамічної відповіді для пралсетинібу охарактеризовані не повністю.
Електрофізіологія серця
Потенціал пралсетинібу щодо подовження інтервалу QT вивчався у 34 пацієнтів з RET-модифікованими солідними пухлинами, які отримували препарат Гаврето в рекомендованих дозах. У дослідженні не спостерігалось значного середнього збільшення QTc (> 20 мс).
Клінічні дослідження
Метастатичний недрібноклітинний рак легень (НДКРЛ) із злиттям RET
Ефективність препарату Гаврето вивчалась у пацієнтів із метастатичним НДКРЛ із злиттям RET у багатоцентровому, нерандомізованому, відкритому, багатокогортному клінічному дослідженні (ARROW). В окремі когорти дослідження були включені пацієнти з метастатичним НДКРЛ із злиттям RET, у яких спостерігалось прогресування захворювання на хіміотерапії на основі препаратів платини, та раніше неліковані пацієнти з метастатичним НДКРЛ. У дослідження були включені пацієнти з безсимптомними метастазами в центральну нервову систему (ЦНС), у тому числі пацієнти зі стабільним застосуванням чи зменшенням застосування кортикостероїдів протягом 2 тижнів до включення в дослідження. Пацієнти отримували препарат Гаврето по 400 мг один раз на день до прогресування захворювання або виникнення неприйнятної токсичності.
Метастатичний НДКРЛ із злиттям RET, раніше лікований за допомогою хіміотерапії на основі платини
Ефективність вивчалась у 130 пацієнтів із НДКРЛ із злиттям RET та вимірюваним захворюванням, які раніше отримували хіміотерапію на основі платини, і були включені в когорту дослідження ARROW. Загальний стан за ECOG становив 0—1 (95 %) або 2 (3,8 %), 99 % пацієнтів мали метастатичне захворювання і 41 % мали в анамнезі або на даний час метастази в ЦНС.
Пацієнти в середньому отримали 2 попередні системні терапії (діапазон 1—6); 42 % отримували попередню анти-PD-1/PD-L1 терапію і 27 % отримували попередню терапію інгібіторами кіназ. Загалом 48 % пацієнтів отримували попередню променеву терапію. Найбільш поширеними партнерами по злиттю RET були KIF5B (70 %) і CCDC6 (19 %).
Таблиця 1
Результати ефективності в дослідженні ARROW (метастатичний НДКРЛ із злиттям RET, раніше лікований за допомогою хіміотерапії на основі платини)
| Показник ефективності | Гаврето (N=130) |
| Частота загальної відповіді (ЧЗВ)a (95 % ДІ) | 63 (54, 71) |
| Повна відповідь, % | 6 |
| Часткова відповідь, % | 57 |
| Тривалість відповіді (ТВ) | (N=82) |
| Медіана, місяці (95 % ДІ) | 38,8 (14,8, NE) |
| Пацієнти з ТВ ≥ 12 місяцівb, % | 66 |
NE — не піддається оцінці.
a Підтверджена частота загальної відповіді за оцінкою BICR.
b Розраховано за допомогою частки пацієнтів, які відповіли на лікування, із спостережуваною тривалістю відповіді протягом щонайменше 6 місяців або більше.
Для 54 пацієнтів, які отримували анти-PD-1 або анти-PD-L1 терапію, послідовно або одночасно з хіміотерапією на основі платини, пошуковий аналіз підгруп ЧЗВ становив 59 % (95 % ДІ: 45, 72) і медіана ТВ становила 22,3 місяця (95 % ДІ: 8,0, NE).
Зі 130 пацієнтів із НДКРЛ із злиттям RET 10 пацієнтів початково мали вимірювані метастази в ЦНС за оцінкою BICR. Жоден пацієнт не отримав променеву терапію на ділянку головного мозку протягом 2 місяців до включення в дослідження. Відповідь із боку внутрішньочерепних уражень спостерігалась у 7 із цих 10 пацієнтів, у тому числі 2 пацієнти з повною відповіддю з боку ЦНС; 71 % пацієнтів, у яких спостерігалась відповідь, ТВ становила ≥ 6 місяців.
Раніше нелікований метастатичний НДКРЛ із злиттям RET
Ефективність вивчалась у 107 пацієнтів з раніше не лікованим метастатичним НДКРЛ із злиттям RET із вимірюваним захворюванням, які були включені в дослідження ARROW.
Загальний стан за ECOG становив 0—1 для 99 % пацієнтів і 98 % пацієнтів мали метастатичне захворювання, 28 % мали в анамнезі або нині метастази в ЦНС. Найбільш поширеними партнерами по злиттю RET були KIF5B (71 %) і CCDC6 (18 %).
Таблиця 2
Результати ефективності в дослідженні ARROW (раніше нелікований метастатичний НДКРЛ із злиттям RET)
| Показник ефективності | Гаврето (N=107) |
| Частота загальної відповіді (ЧЗВ)a (95 % ДІ) | 78 (68, 85) |
| Повна відповідь, % | 7 |
| Часткова відповідь, % | 71 |
| Тривалість відповіді (ТВ) | N=83 |
| Медіана, місяці (95 % ДІ) | 13,4 (9,4, 23,1) |
| Пацієнти з ТВ ≥ 6 місяцівb, % | 45 |
a Підтверджена частота загальної відповіді за оцінкою BICR.
b На підставі спостережуваної тривалості відповіді.
Фармакокінетика.
При пероральному прийомі препарату Гаврето у дозі 400 мг один раз на день натще середнє геометричне в рівноважному стані [% коефіцієнт варіації (CV %)] максимальної спостережуваної концентрації в плазмі крові (Cmax) і площа під кривою концентрація — час (AUC0-24год) пралсетинібу становили 2470 (55,1 %) нг/мл і 36700 (66,3 %) год•нг/мл відповідно. Показники Cmax і AUC пралсетинібу зростали неоднорідно при дозуванні від 60 до 600 мг один раз на день (у 0,15—1,5 раза вище рекомендованої дози).
Концентрація пралсетинібу в плазмі крові досягала рівноважного стану через 3—5 днів. Середній коефіцієнт накопичення був приблизно подвоєним після повторного щоденного перорального прийому один раз на день.
Всмоктування
Медіана часу до досягнення максимальної концентрації (Tmax) варіювала від 2 до 4 годин після прийому разових доз пралсетинібу від 60 до 600 мг.
Вплив їжі
Після прийому однократної дози 200 мг препарату Гаврето з їжею з високим вмістом жиру (приблизно 800—1000 калорій, джерелом 50—60 % з яких були жири), середня (90 % ДІ) Cmax пралсетинібу збільшилась на 104 % (65 %, 153 %), середня (90 % ДІ) AUC0-INF збільшилась на 122 % (96 %, 152 %) і медіана Tmax була відстрочена з 4 до 8,5 години у порівнянні з аналогічними показниками при прийомі препарату натще.
Розподіл
Середній (CV %) очевидний об’єм розподілу (Vd/F) пралсетинібу становить 303 л (68 %). Зв’язування пралсетинібу з білкам становить 97,1% і не залежить від концентрації. Співвідношення вмісту препарату в цільній крові і плазмі крові становить від 0,6 до 0,7.
Елімінація
Середній (± стандартне відхилення) період напіввиведення (T½) пралсетинібу із плазми крові становить 15,7 години (9,8) після прийому однократних доз і 20 годин (11,7) після прийому багатократних доз пралсетинібу. Середній (CV %) очевидний загальний кліренс (CL/F) пралсетинібу становив 10,9 л/годину (66 %) у рівноважному стані.
Метаболізм
Пралсетиніб в основному метаболізується CYP3A4 і меншою мірою — CYP2D6 і CYP1A2, in vitro. Після перорального прийому однократної дози 310 мг міченого радіоізотопом пралсетинібу здоровими особами метаболіти пралсетинібу у результаті окислення та глюкуронізації були виявлені на рівні 5 % або менше.
Екскреція
Приблизно 73 % (66 % у незміненому вигляді) сумарної пероральної дози міченого радіоізотопом [14C] пралсетинібу виділилось з фекаліями і 6 % (4,8 % у незміненому вигляді) — із сечею.
Особливі групи пацієнтів
Не спостерігалось клінічно значимої різниці у ФК пралсетинібу залежно від віку (від 19 до 87 років), статі, раси (370 — європеоїдної раси, 22 — негроїдної раси і 61 — азійського походження) і маси тіла (від 32,1 до 128 кг) пацієнта. Порушення функції нирок легкого та помірного ступеня тяжкості (кліренс креатиніну 30—89 мл/хв) не впливало на експозицію пралсетинібу. Не вивчалося застосування пралсетинібу пацієнтам із тяжким порушенням функції нирок (кліренс креатиніну < 15 мл/хв).
Порушення функції печінки
Порушення функції печінки легкого ступеня (рівень загального білірубіну ≤ 1,0 × ВМН і АСТ > ВМН або рівень загального білірубіну від > 1,0 до 1,5 × ВМН і будь-який рівень АСТ) не впливало на ФК пралсетинібу. Не вивчалося застосування пралсетинібу пацієнтам із помірним (рівень загального білірубіну від > 1,5 до 3,0 × ВМН і будь-який рівень АСТ) або тяжким (рівень загального білірубіну > 3,0 ВМН і будь-який рівень АСТ) порушенням функції печінки.
Дослідження взаємодії з іншими лікарськими засобами
Клінічні дослідження і модель-орієнтований підхід
Інгібітори CYP3A. Супутнє застосування багаторазових доз інгібіторів CYP3A призводить до збільшення Cmax та AUC пралсетинібу.
Таблиця 3
Спостережуване і прогнозоване збільшення експозиції пралсетинібу після супутнього застосування з інгібіторами CYP3A
| Тип інгібітору | Супутнє застосування з інгібітором CYP3A | Збільшення Cmax пралсетинібу | Збільшення AUC пралсетинібу |
| Спостережувані | |||
| Інгібітор P-gp і сильний інгібітор CYP3A | Ітраконазол (200 мг двічі на день в день 1 із наступним застосуванням 200 мг один раз на день) | 84 % | 251 % |
| Прогнозовані | |||
| Сильні інгібітори CYP3A | Вориконазол (400 мг двічі на день в день 1 із наступним застосуванням 200 мг двічі на день) | 20 % | 122 % |
| Супутнє застосування інгібітору P-gp і помірного інгібітору CYP3A | Верапаміл (80 мг тричі на день) | 60 % | 108 % |
| Помірні інгібітори CYP3A | Флуконазол (400 мг один раз на день) | 15 % | 71 % |
Інгібітори P-gp. Супутнє застосування циклоспорину (однократна доза 600 мг) із однократною дозою 200 мг пралсетинібу супроводжувалось збільшенням AUC пралсетинібу на 81 % і Cmax — на 48 % порівняно із застосуванням лише 200 мг пралсетинібу.
Сильні індуктори CYP3A. Супутнє застосування 600 мг рифампіну один раз на день із однократною дозою 400 мг препарату Гаврето супроводжувалось зниженням AUC0-INF пралсетинібу на 68 % і Cmax — на 30 %.
Помірні індуктори CYP3A. Супутнє застосування багаторазових доз ефавіренцу (600 мг один раз на день) призводить до прогнозованого зменшення AUC пралсетинібу на 45 % і Cmax — на 18 %.
Слабкі індуктори CYP3A. Не спостерігались клінічно значимі зміни ФК пралсетинібу при супутньому застосуванні препарату Гаврето та слабких індукторів CYP3A.
Антацидні засоби. Не спостерігались клінічно значимі зміни ФК пралсетинібу при супутньому застосуванні препарату Гаврето та засобів, що зменшують кислотність шлунка.
Дослідження іn vitro
Ферменти системи цитохрому P450 (CYP). Пралсетиніб є залежним від часу інгібітором CYP3A4/5 та інгібітором CYP2C8, CYP2C9 і CYP3A4/5, однак не є інгібітором CYP1A2, CYP2B6, CYP2C19 або CYP2D6 в клінічно значимих концентраціях.
Пралсетиніб є індуктором CYP2C8, CYP2C9 і CYP3A4/5, однак не є індуктором СYP1A2, CYP2B6 або CYP2C19 в клінічно значимих концентраціях.
Системи транспортерів. Пралсетиніб є субстратом P-глікопротеїну (P-gp) і білка резистентності раку молочної залози (BCRP), однак не є субстратом ефлюксної помпи жовчних кислот (BSEP), білка-транспортера органічних аніонів [OCT]1, OCT2, поліпептидів, що транспортують органічні аніони [OATP]1B1, OATP1B3, білка множинної резистентності і виведення токсинів [MATE]1, MATE2-K, транспортерів органічних аніонів [OAT]1 або OAT3.
Пралсетиніб є інгібітором P-gp, BCRP, OATP1B1, OATP1B3, OAT1, MATE1, MATE2-K і BSEP, однак не є інгібітором OCT1, OCT2 і OAT1A3 в клінічно значимих концентраціях.
Метастатичний недрібноклітинний рак легень (НДКРЛ), позитивний на злиття RET
Лікування дорослих пацієнтів з НДКРЛ, позитивним на злиття RET.
Підвищена чутливість до пралсетинібу або до будь-якої допоміжної речовини, що входить до складу препарату.
Вплив інших лікарських засобів на препарат Гаврето
Сильні або помірні інгібітори CYP3A та/або інгібітори P-gp
Супутнє застосування із сильними або помірними інгібіторами CYP3A та/або інгібіторами P-gp збільшує експозицію пралсетинібу, що може призвести до збільшення ризику виникнення побічних реакцій, пов’язаних із застосуванням препарату Гаврето. Потрібно уникати супутнього застосування препарату Гаврето із сильним або помірним інгібітором CYP3A та/або інгібітором P-gp. При неможливості уникнення супутнього застосування з будь-яким зазначеним вище інгібітором дозу препарату Гаврето потрібно зменшити (див. розділи «Спосіб застосування та дози», «Фармакокінетика»).
Сильні або помірні індуктори CYP3A
Супутнє застосування препарату Гаврето із сильним або помірним індуктором CYP3A призводить до зменшення експозиції пралсетинібу, що може призвести до зниження ефективності препарату Гаврето. Потрібно уникати супутнього застосування препарату Гаврето із сильними або помірними індукторами CYP3A. При неможливості уникнення супутнього застосування із сильними або помірними індукторами CYP3A дозу препарату Гаврето потрібно збільшити (див. розділи «Спосіб застосування та дози», «Фармакокінетика»).
Інтерстиціальне захворювання легень (ІЗЛ)/пневмоніт
У пацієнтів, які отримують лікування препаратом Гаврето, можуть з’являтися тяжкі, загрозливі для життя і летальні випадки ІЗЛ/пневмоніту. Пневмоніт виникав у 12 % пацієнтів, які отримували препарат Гаврето, у тому числі у 3,3 % — ступеня 3—4 і у 0,2 % — летальні реакції.
Потрібно спостерігати за станом пацієнтів щодо появи симптомів із боку легень, що свідчать про ІЗЛ/пневмоніт. Лікування препаратом Гаврето потрібно призупинити і негайно обстежити кожного пацієнта щодо ІЗЛ при виникненні або погіршенні респіраторних симптомів, які можуть свідчити про ІЗЛ (таких як задишка, кашель і лихоманка). Залежно від тяжкості підтвердженого ІЗЛ призупиняють застосування, зменшують дозу або назавжди відміняють лікування препаратом Гаврето (див. розділ «Спосіб застосування та дози»).
Артеріальна гіпертензія
Артеріальна гіпертензія виникала у 35 % пацієнтів, включаючи артеріальну гіпертензію ступеня 3 у 18 % пацієнтів (див. розділ «Небажані реакції»). Загалом 8 % пацієнтів було необхідне переривання дозування і 4,8 % пацієнтам — зниження дози через артеріальну гіпертензію. Артеріальна гіпертензія, що виникала у період лікування, найчастіше коригувалася за допомогою антигіпертензивного лікування.
Не можна починати застосування препарату Гаврето пацієнтам із неконтрольованою гіпертензією. Потрібно оптимізувати артеріальний тиск до початку лікування препаратом Гаврето. Моніторувати рівень артеріального тиску через 1 тиждень, надалі — щонайменше щомісячно, а також за клінічними показаннями. У разі потрібності розпочинають або корегують антигіпертензивну терапію. Залежно від тяжкості призупиняють застосування, зменшують дозу або назавжди відміняють лікування препаратом Гаврето (див. розділ «Спосіб застосування та дози»).
Гепатотоксичність
Серйозні небажані реакції з боку печінки виникали у 1,5 % пацієнтів, які отримували лікування препаратом Гаврето. Підвищення рівня АСT спостерігалось у 49 % пацієнтів, у тому числі ступеня 3 або 4 у 7 %, і підвищення рівня АЛT — у 37 % пацієнтів, включаючи 3 або 4 ступеня тяжкості у 4,8 % пацієнтів (див. розділ «Небажані реакції»). Медіана часу до першого збільшення рівня АСT становила 15 днів (діапазон: від 5 днів до 2,5 року) і для АСТ — 24 дні (діапазон: від 7 днів до 3,7 року).
Потрібно моніторувати рівень АСT та АЛT до початку лікування препаратом Гаврето, кожні 2 тижні під час перших 3 місяців, і в подальшому щомісячно та за клінічними показаннями. Залежно від тяжкості призупиняють застосування, зменшують дозу або назавжди відміняють лікування препаратом Гаврето (див. розділ «Спосіб застосування та дози»).
Геморагічні явища
При лікуванні препаратом Гаврето можуть з’являтися серйозні, у тому числі летальні, геморагічні явища. Геморагічні явища ≥ 3 ступеня тяжкості виникали у 4,1 % пацієнтів, які отримували лікування препаратом Гаврето, у тому числі один пацієнт із летальним геморагічним явищем.
Пацієнтам із тяжкими чи такими, що загрожують життю, кровотечами лікування препаратом Гаврето слід відмінити назавжди (див. розділ «Спосіб застосування та дози»).
Подовження інтервалу QT
Подовження інтервалу QT спостерігалось у пацієнтів, які отримували препарат Гаврето в клінічних дослідженнях (див. підрозділ “Електрофізіологія серця”). Тому до початку лікування препаратом Гаврето інтервал QTc має становити ≤470 мс, рівень електролітів та ТТГ мають бути в межах норми. Перед та у період лікування препаратом Гаврето потрібно відкоригувати гіпокаліємію, гіпомагніємію та гіпокальціємію.
Електрокардіограму (ЕКГ) та рівень електролітів в сироватці крові потрібно контролювати в кінці першого тижня та першого місяця лікування препаратом Гаврето, після цього періодично за клінічними показами і також в залежності від наявності інших факторів ризику (зокрема, інтеркурентна діарея, блювання, нудота, супутній прийом інших лікарських засобів).
Пралсетиніб потрібно застосовувати обережно у пацієнтів із аритмією серця чи подовження інтервалу QT в медичному анамнезі, a також у пацієнтів, які отримують сильні інгібітори CYP 3A4 або препарати, які асоціюються із подовженням QT/QTc. Потрібно контролювати пацієнтів із суттєвим ризиком розвитку подовження QTc, включаючи пацієнтів із відомим синдромом подовження інтервалу QT, клінічно значимими брадиаритміями і тяжкою чи неконтрольованою серцевою недостатністю. Потрібно контролювати інтервал QT частіше при супутньому застосуванні пралсетинібу із сильними чи помірними інгібіторами CYP3A або лікарськими засобами, які подовжують інтервал QTc. Пралсетиніб не вивчався у пацієнтів із клінічно значимим активним серцево-судинним захворюванням або нещодавнім інфарктом міокарда.
Можливо буде потрібно переривання, модифікація дози або відміна препарату Гаврето (див. розділ “Спосіб застосування та дози”).
Синдром лізису пухлини
У пацієнтів із медулярною карциномою щитоподібної залози, які отримували лікування препаратом Гаврето, повідомлялись випадки синдрому лізису пухлини (див. розділ «Небажані реакції»). Ризик виникнення синдрому лізису пухлини може спостерігатись у пацієнтів зі швидко зростаючими пухлинами, високим пухлинним навантаженням, порушенням функції нирок або дегідратацією.
Потрібно спостерігати за станом пацієнтів із факторами ризику, розглянути питання про відповідну профілактику, включаючи гідратацію, і за клінічними показаннями призначити лікування.
Ризик порушення загоєння ран
Порушення загоєння ран можливе у пацієнтів, які отримують препарати, що інгібують шлях передачі сигналу, опосередкований судинним ендотеліальним фактором росту (VEGF). Тому препарат Гаврето може несприятливо впливати на загоєння ран.
Потрібно відмінити прийом препарату Гаврето щонайменше за 5 днів до планового хірургічного втручання. Не можна застосовувати препарат щонайменше 2 тижні після великого оперативного втручання і до належного загоєння рани. Безпека продовження лікування препаратом Гаврето після зникнення ускладнень загоєння рани не встановлена.
Ембріофетальна токсичність
За результатами досліджень на тваринах та з огляду на механізм дії, препарат Гаврето може спричиняти шкоду для плода при застосуванні вагітним жінкам. Пероральне введення пралсетинібу вагітним самкам щурів під час періоду органогенезу призводило до вад розвитку та ембріолетальності при експозиції у самок, меншій ніж експозиція у людини при застосуванні препарату в клінічній дозі 400 мг один раз на день.
Потрібно проінформувати вагітних жінок про потенційний ризик для плода та порадити жінкам репродуктивного віку застосовувати ефективні негормональні методи контрацепції у період лікування препаратом Гаврето і протягом 2 тижнів після отримання останньої дози. Потрібно порадити чоловікам та їхнім партнеркам — жінкам репродуктивного віку застосовувати ефективні методи контрацепції у період лікування препаратом Гаврето і протягом 1 тижня після отримання останньої дози (див. розділ «Застосування у період вагітності або годування груддю»).
Дисплазія зон росту
Потрібно контролювати зони росту у дітей з відкритими зонами росту. Потрібно розглянути питання про переривання або припинення лікування з огляду на ступінь тяжкості будь-яких відхилень зон росту та за результатами оцінки співвідношення користь-ризик для кожного окремого пацієнта.
Супутні альтерації драйверних онкогенів
Ефективність та безпека застосування препарату Гаврето для пацієнтів із супутніми альтераціями драйверних онкогенів не встановлені. Такі таргетні драйверні альтерації, що підлягають лікуванню, були виключені із дослідження BLU-667-1101: НДКРЛ — із таргетними альтераціями EGFR, ALK, ROS1 або BRAF, що підлягають лікуванню.
Вміст натрію
Цей препарат містить менше 1 ммоль (23 мг)/капсулу натрію, тобто практично вільний від натрію.
Застосування у період вагітності або годування груддю.
Вагітність
Резюме ризику
За даними досліджень на тваринах та з огляду на механізм дії, препарат Гаврето може спричиняти шкоду для плода при застосуванні вагітним жінкам (див. розділ «Фармакодинаміка»). Немає даних для інформування вагітних жінок про ризики, пов’язані із застосуванням препарату Гаврето. Пероральне введення пралсетинібу вагітним самкам щурів під час періоду органогенезу призводило до вад розвитку та ембріолетальності при експозиції у самок, меншій, ніж експозиція у людини при застосуванні в клінічній дозі 400 мг один раз на день (див. підрозділ «Дані»). Потрібно проінформувати вагітних жінок про потенційний ризик для плода.
У загальній популяції США розрахунковий базовий ризик суттєвих вроджених вад та невиношування вагітності в клінічно визнаних випадках вагітності становить 2—4 % і 15—20 % відповідно.
Дані
Дані, отримані у дослідженнях на тваринах
В ембріофетальному онтогенетичному дослідженні пероральне щоденне введення пралсетинібу в дозі ≥ 20 мг/кг (експозиція приблизно у 1,8 раза перевищувала експозицію у людини на основі площі під кривою [AUC] при застосуванні в клінічній дозі 400 мг) один раз на день вагітним самкам щурів під час періоду органогенезу призводило до 100 % постімплантаційної загибелі плода. Постімплантаційна загибель плода також відбулась при застосуванні у дозі 10 мг/кг (приблизно 0,6-кратний рівень експозиції у людини на основі AUC при застосуванні в клінічній дозі 400 мг). Застосування пралсетинібу один раз на день в дозі ≥ 5 мг/кг (приблизно 0,2-кратний рівень експозиції у людини на основі AUC при застосуванні в клінічній дозі 400 мг) призводило до збільшення частоти вісцеральних вад розвитку і відхилень (відсутні або невеликі нирки і сечовід, відсутній ріг матки, неправильне розташування нирок або яєчок, ретроезофагеальна дуга аорти), а також вад і відхилень із боку скелета (аномалії хребта та ребер і зменшення окостеніння).
Лактація
Резюме ризику
Немає інформації щодо присутності пралсетинібу або його метаболітів в грудному молоці людини або впливу на дитину, яка знаходиться на грудному вигодовуванні, або на вироблення молока. Зважаючи на ризик виникнення серйозних побічних реакцій у дітей, які знаходяться на грудному вигодовуванні, потрібно порадити жінкам не годувати груддю у період лікування препаратом Гаврето та протягом 1 тижня після отримання останньої дози.
Жінки і чоловіки репродуктивного віку
На підставі даних, отриманих у дослідженнях на тваринах, препарат Гаврето може спричиняти ембріолетальність та вроджені вади при експозиції, меншій, ніж експозиція у людини при застосуванні в клінічній дозі 400 мг щоденно (див. підрозділ «Вагітність»).
Тестування на вагітність
До початку лікування препаратом Гаврето потрібно визначити статус вагітності у жінок репродуктивного віку (див. підрозділ «Вагітність»).
Контрацепція
При застосуванні під час вагітності препарат Гаврето може спричиняти шкоду для плода (див. підрозділ «Вагітність»).
Жінки
Потрібно порадити жінкам репродуктивного віку застосовувати ефективні негормональні методи контрацепції у період лікування препаратом Гаврето і протягом 2 тижнів після отримання останньої дози. Препарат Гаврето може зробити гормональні контрацептиви неефективними.
Чоловіки
Потрібно порадити чоловікам та їхнім партнеркам — жінкам репродуктивного віку застосовувати ефективні методи контрацепції у період лікування препаратом Гаврето і протягом 1 тижня після отримання останньої дози.
Безпліддя
На підставі патогістологічних даних, отриманих з тканин репродуктивних органів у самців та самок щурів, а також на підставі результатів спеціального дослідження впливу на фертильність, в якому тварини обох статей отримували препарат і були спарені між собою, препарат Гаврето може порушувати фертильність
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Дослідження впливу препарату Гаврето на швидкість реакції при керуванні автотранспортом або іншими механізмами не проводились. Слід дотримуватися обережності під час керування автотранспортом і роботи з іншими механізмами, оскільки під час застосування препарату Гаврето може виникати слабкість та інші небажані реакції (див. розділ «Небажані реакції»).
Вибір пацієнта
Пацієнтів для лікування препаратом Гаврето обирають на підставі наявності злиття гена RET (НДКРЛ).
Рекомендоване дозування
Рекомендована доза препарату Гаврето становить 400 мг перорально один раз на день натще (без прийому їжі протягом принаймні 2 годин до прийому препарату Гаврето та принаймні однієї години після прийому препарату Гаврето) (див. розділ «Фармакокінетика»). Лікування слід продовжувати до прогресування захворювання або виникнення неприйнятної токсичності.
Якщо дозу препарату Гаврето пропущено, її можна прийняти якомога швидше того ж дня. Відновити регулярний щоденний графік прийому препарату Гаврето на наступний день.
Не слід приймати додаткову дозу при блюванні після прийому препарату Гаврето і прийняти наступну дозу відповідно до режиму дозування.
Модифікація дози через небажані реакції
Рекомендації щодо зниження дози та модифікації дози через небажані реакції наведено в таблицях 4—5.
Таблиця 4
Рекомендації щодо зниження дози препарату Гаврето через небажані реакції
| Зниження дози | Рекомендована доза |
| Перше | 300 мг один раз на день |
| Друге | 200 мг один раз на день |
| Третє | 100 мг один раз на день |
Препарат Гаврето слід відмінити назавжди пацієнтам із непереносимістю дози 100 мг перорально один раз на день.
Таблиця 5
Рекомендації щодо модифікації дози препарату Гаврето через небажані реакції
| Побічна реакція | Тяжкість* | Модифікація дози |
| Інтерстиціальне захворювання легень (ІЗЛ)/пневмоніт (див. розділ «Особливості застосування») | Ступінь 1 або 2 | Призупинити прийом Гаврето до відновлення стану. Продовжити лікування зниженою дозою відповідно до таблиці 1. Припинити застосування Гаврето назавжди у разі рецидивуючого ІЗЛ/пневмоніту. |
| Ступінь 3 або 4 | Припинити застосування Гаврето назавжди у разі підтвердженого ІЗЛ/пневмоніту. | |
| Артеріальна гіпертензія (див. розділ «Особливості застосування») | Ступінь 3 | Призупинити прийом Гаврето при артеріальній гіпертензії ступеня 3, що персистує незважаючи на оптимальну антигіпертензивну терапію. Продовжити лікування зниженою дозою після досягнення контролю гіпертензії. |
| Ступінь 4 | Відмінити препарат Гаврето. | |
| Гепатотоксичність (див. розділ «Особливості застосування») | Ступінь 3 або 4 | Призупинити прийом Гаврето і моніторувати рівень АСT/АЛT один раз на тиждень до зменшення тяжкості до ступеня l або до вихідного рівня. Продовжити лікування зниженою дозою (таблиця 1). Припинити застосування Гаврето назавжди у разі рецидиву гепатотоксичності ступеня 3 або вище. |
| Геморагічні явища (див. розділ «Особливості застосування») | Ступінь 3 або 4 | Призупинити прийом Гаврето до відновлення до вихідного стану або ступеня 0 або 1. Припинити застосування Гаврето назавжди у разі тяжких або таких, що загрожують життю, геморагічних явищ. |
| Подовження інтервалу QT | Ступінь 3 | Призупинити прийом Гаврето при інтервалі QTc >500 мс до відновлення показника інтервалу QTc до <470 мс. Продовжити лікування в тій же дозі у разі ідентифікації та корекції факторів ризику, що спричиняють подовження інтервалу QT. Продовжити лікування зниженою дозою, якщо не ідентифіковані інші фактори ризику, які спричиняють подовження інтервалу QT. |
| Ступінь 4 | Припинити застосування Гаврето назавжди у разі аритмії, що загрожує життю. | |
| Інші небажані реакції (див. розділ «Особливості застосування») | Ступінь 3 або 4 | Призупинити прийом Гаврето до покращення стану до ≤ ступеня 2. Продовжити лікування зниженою дозою (таблиця 1). Припинити застосування Гаврето назавжди у разі рецидивуючих побічних реакцій ступеня 4. |
* Тяжкість визначена на підставі загальних термінологічних критеріїв небажаних явищ Національного інституту раку (NCI CTCAE) версії 4.03.
Модифікація дози при застосуванні в комбінації з інгібіторами CYP3A та/або інгібіторами P-глікопротеїну (P-gp)
Потрібно уникати супутнього застосування препарату Гаврето із будь-якими із нижчезазначених речовин:
Якщо неможливо уникнути супутнього застосування із будь-яким зазначеним вище інгібітором, поточну дозу препарату Гаврето потрібно знизити відповідно до таблиці 6. Коли після відміни інгібітору мине час, що дорівнює 3—5 періодам напіввиведення інгібітору, прийом препарату Гаврето відновлюють у дозі, що застосовувалась до початку прийому інгібітору (див. розділи «Взаємодія з іншими лікарськими засобами та інші види взаємодій», «Фармакокінетика»).
Таблиця 6
Рекомендації щодо модифікації дози препарату Гаврето при супутньому застосуванні з інгібіторами CYP3A та/або інгібіторами P-gp
| Поточна доза препарату Гаврето | Рекомендована доза препарату Гаврето при супутньому застосуванні із: | |
| інгібіторами P-gp та сильними інгібіторами CYP3A, що застосовуються супутньо |
| |
| 400 мг перорально один раз на день | 200 мг перорально один раз на день | 300 мг перорально один раз на день |
| 300 мг перорально один раз на день | 200 мг перорально один раз на день | 200 мг перорально один раз на день |
| 200 мг перорально один раз на день | 100 мг перорально один раз на день | 100 мг перорально один раз на день |
Модифікація дози при супутньому застосуванні із сильними індукторами CYP3A
Потрібно уникати супутнього застосування препарату Гаврето із будь-якими із нижчезазначених речовин:
Якщо неможливо уникнути супутнього застосування із будь-яким зазначеним вище індуктором, поточну дозу препарату Гаврето потрібно збільшити відповідно до рекомендацій, викладених у таблиці 7, починаючи з 7 дня супутнього застосування препарату Гаврето із індуктором. Через щонайменше 14 днів після відміни індуктора відновлюють прийом препарату Гаврето у дозі, що застосовувалась до початку застосування індуктора (див. розділи «Взаємодія з іншими лікарськими засобами та інші види взаємодій», «Фармакокінетика»).
Таблиця 7
Рекомендації щодо модифікації дози препарату Гаврето при супутньому застосуванні з індукторами CYP3A
| Поточна доза препарату Гаврето | Рекомендована доза препарату Гаврето при супутньому застосуванні із: | |
| сильними індукторами CYP3A | помірними індукторами CYP3A | |
| 400 мг перорально один раз на день | 800 мг перорально один раз на день | 600 мг перорально один раз на день |
| 300 мг перорально один раз на день | 600 мг перорально один раз на день | 500 мг перорально один раз на день |
| 200 мг перорально один раз на день | 400 мг перорально один раз на день | 300 мг перорально один раз на день |
Особливі групи пацієнтів
Порушення функції печінки
Не вивчалося застосування препарату Гаврето пацієнтам із помірним порушенням функції печінки (рівень загального білірубіну від > 1,5 до 3,0 × ВМН [верхня межа норми] і будь-який рівень АСТ) або тяжким порушенням функції печінки (рівень загального білірубіну > 3,0 × ВМН і будь-який рівень АСТ). Пацієнтам із порушенням функції печінки легкого ступеня (рівень загального білірубіну ≤ ВМН і АСT > ВМН або рівень загального білірубіну від > 1 до 1,5 × ВМН і будь-який рівень АСТ) корекція дози не потрібна (див. розділ «Фармакокінетика»).
Пацієнти літнього віку
Із 540 учасників дослідження ARROW, які отримували препарат Гаврето в рекомендованій дозі 400 мг один раз на день, 31 % пацієнтів були віком від 65 років, причому 7 % пацієнтів були віком від 75 років.
Загалом не спостерігалось відмінностей у фармакокінетиці (ФK), безпеці чи ефективності застосування препарату пацієнтам віком від 65 років у порівнянні з пацієнтами молодшого віку.
Діти
Безпека та ефективність застосування препарату Гаврето не була встановлена для дітей із НДКРЛ із злиттям RET.
Токсикологічні дані у дослідженнях на тваринах
У 4-тижневому дослідженні токсичності при застосуванні в повторних дозах у приматів (тварин) спостерігалась дисплазія зони росту в стегновій кістці при застосуванні в дозах, що призводять до експозиції, подібній до експозиції у людини (AUC) при прийомі в клінічній дозі 400 мг. У щурів спостерігалось збільшення товщини зони росту в стегновій кістці і груднині, а також аномалії зубів (різці) (переломи, зміни матриці дентину, дегенерація амелобластів/одонтобластів, некроз) в 4- та 13-тижневих дослідженнях при застосуванні у дозах, що призводять до експозиції, подібній до експозиції у людини (AUC) при прийомі в клінічній дозі 400 мг. Відновлення не оцінювалось у 13-тижневому токсикологічному дослідженні, однак не спостерігалось повного відновлення товщини зони росту в стегновій кістці та дегенерації різців у 28-денному дослідженні у щурів.
Потрібно моніторувати зони росту у підлітків із відкритими зонами росту. Слід розглянути питання про переривання або припинення терапії залежно від тяжкості будь-якої аномалії зони росту і на підставі оцінки користь/ризик для кожного пацієнта.
Немає досвіду передозування людини препаратом Гаврето в клінічних дослідженнях. У разі передозування потрібно ретельно контролювати стан пацієнта та проводити загальні підтримувальні заходи. Специфічного антидоту у разі передозування препаратом Гаврето немає.
Досвід клінічних досліджень
Оскільки клінічні дослідження проводяться в різних умовах, частота побічних реакцій, яка спостерігалася в клінічних дослідженнях препарату, не може безпосередньо порівнюватися з частотою в клінічних дослідженнях іншого препарату і може не відображати частоту, що спостерігалася на практиці.
Сукупна популяція безпеки в розділі «Особливості застосування» відображає експозицію препарату Гаврето як монотерапії при застосуванні в дозі 400 мг один раз на день перорально у 540 пацієнтів в дослідженні ARROWІз 540 пацієнтів, які отримували препарат Гаврето, 71 % приймали препарат протягом 6 місяців або довше і 57 % — довше одного року. Найбільш поширеними побічними реакціями (≥ 25 %) були кістково-м’язовий біль, запор, артеріальна гіпертензія, діарея, слабкість, набряки, підвищення температури тіла, кашель. Найбільш поширеними відхиленнями лабораторних показників ступеня 3—4 (≥ 2 %) були зменшення числа лімфоцитів, нейтрофілів, зниження рівня гемоглобіну, фосфату, зменшення числа лейкоцитів, зменшення рівня натрію, збільшення рівня аспартатамінотрансферази (АСT), аланінамінотрансферази (АЛT), зменшення рівня кальцію (скоригованого), зменшення числа тромбоцитів, збільшення рівня лужної фосфатази, збільшення рівня калію, зниження рівня калію та підвищення рівня білірубіну.
Недрібноклітинний рак легень, позитивний на злиття RET
Вивчалась безпека застосування препарату Гаврето як монотерапії в дозі 400 мг один раз на день перорально за участю 281 пацієнта з метастатичним реаранжованим під час трансфекції (позитивний на злиття RET) НДКРЛ в дослідженні ARROWІз 281 пацієнта, які отримували препарат Гаврето, тривалість експозиції у 72 % пацієнтів становила 6 місяців або більше і у 56 % становила ≥ 1 року.
Медіана віку становила 60 років (діапазон: 26—87 років); 54 % становили жінки, 46 % були європеоїдної раси, 46 % — азійського походження і 4 % — латино-американці.
Серйозні небажані реакції виникали у 65 % пацієнтів, які отримували препарат Гаврето. Найбільш поширеними серйозними побічними реакціями (у ≥ 2 % пацієнтів) були пневмонія, анемія, пневмоніт, підвищення температури тіла, сепсис, інфекції сечовивідних шляхів, коронавірусна інфекція, плевральний випіт, задишка, м’язово-скелетний біль, легенева емболія і судоми. Побічна реакція з летальним наслідком виникала у 7 % пацієнтів; побічна реакція з летальним наслідком, яка виникла у > 1 пацієнта, включала пневмонію (n = 8), сепсис (n = 3) та COVID (n = 3).
Препарат Гаврето був відмінений назавжди через побічну реакцію 20 % пацієнтів. Небажані реакції, які призвели до передчасної відміни лікування назавжди і які виникали у ≥ 2 пацієнтів, включали пневмоніт (3,2 %) та пневмонію (2,8 %).
Переривання дозування через побічну реакцію було необхідне 73 % пацієнтів, які отримували препарат Гаврето. Небажані реакції, через які було необхідне припинення дозування ≥ 2 % пацієнтів, включали анемію, пневмонію, пневмоніт, нейтропенію, артеріальну гіпертензію, підвищення рівня креатинфосфокінази в крові, слабкість, підвищення температури тіла, збільшення рівня АСT, коронавірусу інфекцію, діарею, гіпофосфатемію, м’язово-скелетний біль, тромбоцитопенію, задишку, кровотечу, лейкопенію, лімфопенію, набряки, сепсис і блювання.
Зменшення дози через побічну реакцію було необхідне 51 % пацієнтів, які отримували препарат Гаврето. Небажані реакції, через які було необхідне зниження дози ≥ 2 % пацієнтів, включали анемію, нейтропенію, пневмоніт, підвищення рівня креатинфосфокінази в крові, лейкопенію, артеріальну гіпертензію, слабкість, пневмонію і лімфопенію.
У таблиці 8 наведено резюме побічних реакцій, які виникали у пацієнтів із НДКРЛ, позитивним на злиття RET, у дослідженні АRROW.
Таблиця 8
Небажані реакції (≥ 15 %) у пацієнтів із НДКРЛ, позитивним на злиття RET, які отримували препарат Гаврето в дослідженні ARROW
| Небажані реакції | Гаврето N = 281 | |
| Ступеня 1—4 (%) | Ступеня 3 або 4 (%) | |
| Розлади з боку шлунково-кишкового тракту | ||
| Запор | 45 | 0,7 |
| Діарея | 30 | 2,5 |
| Нудота | 19 | 0 |
| Сухість у роті | 17 | 0 |
| Загальні розлади та розлади у місці введення | ||
| Набряк1 | 44 | 0 |
| Слабкість2 | 42 | 2,5 |
| Підвищення температури тіла | 29 | 0,7 |
| Розлади з боку опорно-рухової системи | ||
| Кістково-м’язовий біль3 | 44 | 2,5 |
| Підвищення рівня креатинфосфокінази в крові | 19 | 9 |
| Судинні розлади | ||
| Артеріальна гіпертензія4 | 38 | 18 |
| Розлади з боку дихальної системи, органів грудної клітки та середостіння | ||
| Кашель5 | 36 | 0,4 |
| Задишка | 21 | 2,1 |
| Інфекції та інвазії | ||
| Пневмонія6 | 24 | 13 |
| Інфекції сечовивідних шляхів | 16 | 3,6 |
| Порушення обміну речовин і харчування | ||
| Зниження апетиту | 18 | 1,1 |
| Розлади з боку нервової системи | ||
| Розлади смаку7 | 17 | 0 |
| Головний біль8 | 15 | 1,1 |
| Порушення з боку шкіри і підшкірної клітковини | ||
| Висипання9 | 17 | 0 |
1 Включає такі терміни переважного використання: набряк, набряклість обличчя, периферична набряклість, генералізований набряк, периферичний набряк, набряк обличчя, періорбітальний набряк,
набряк повік, набряклість, локалізований набряк.
2 Включає такі терміни переважного використання: слабкість, астенія.
3 Включає такі терміни переважного використання: міалгія, артралгія, біль у кінцівках, біль у шиї, кістково-м’язовий біль, біль у спині, кістково-м’язовий біль у грудній клітці, біль у кістках, кістково-м’язова скутість.
4 Включає такі терміни переважного використання: артеріальна гіпертензія, підвищення рівня артеріального тиску.
5 Включає такі терміни переважного використання: кашель, продуктивний кашель, синдром кашлю верхніх дихальних шляхів.
6 Включає такі терміни переважного використання: пневмонія, пневмоцистна пневмонія, цитомегаловірусна пневмонія, атипова пневмонія, інфекція легень, бактеріальна пневмонія, гемофільна пневмонія, грипозна пневмонія, стрептококова пневмонія, вірусна пневмонія, псевдомонадна пневмонія.
7 Включає такі терміни переважного використання: дисгевзія, агевзія.
8 Включає такі терміни переважного використання: головний біль, головний біль напруги.
9 Включає такі терміни переважного використання: висипання, макулопапульозне висипання, акнеформний дерматит, еритема, генералізоване висипання, папульозне висипання, макулярне висипання, еритематозне висипання.
Клінічно значимі небажані реакції, які виникали < 15 % пацієнтів, включали пневмоніт (14 %),
блювання (14 %), біль у животі (14 %) і стоматит (6 %).
У таблиці 9 наведено резюме відхилень лабораторних показників у дослідженні ARROW.
Таблиця 9
Окремі відхилення лабораторних показників (≥ 20 %), які погіршились від вихідного рівня у пацієнтів із НДКРЛ, позитивним на злиття RET, які отримували препарат Гаврето в дослідженні ARROW
| Відхилення лабораторних показників | Гаврето N = 281 | |
| Ступеня 1—4 (%) | Ступеня 3—4 (%) | |
| Біохімічні показники | ||
| Підвищення рівня АСТ | 80 | 3,2 |
| Підвищення рівня АЛТ | 58 | 3,9 |
| Зниження рівня альбуміну | 52 | 0 |
| Зниження рівня кальцію (скоригованого) | 50 | 1,8 |
| Зниження рівня фосфату | 50 | 17 |
| Підвищення рівня креатиніну | 45 | 1,4 |
| Підвищення рівня лужної фосфатази | 43 | 2,5 |
| Зниження рівня натрію | 42 | 10 |
| Зниження рівня калію | 27 | 4,6 |
| Підвищення рівня калію | 27 | 1,8 |
| Зниження рівня магнію | 25 | 0 |
| Підвищення рівня білірубіну | 20 | 1,8 |
| Гематологічні показники | ||
| Зменшення числа лейкоцитів | 79 | 11 |
| Зниження рівня гемоглобіну | 78 | 18 |
| Зменшення числа лімфоцитів | 73 | 32 |
| Зменшення числа нейтрофілів | 70 | 21 |
| Зменшення числа тромбоцитів | 33 | 5 |
Клінічно значимі відхилення лабораторних показників, які виникали у < 20 % пацієнтів, які отримували препарат Гаврето, включали збільшення рівня магнію (14 %).
Повідомлення про небажані реакції після реєстрації препарату має важливе значення. Це дає змогу проводити моніторинг співвідношення користь/ризик при застосуванні цього препарату. Медичним та фармацевтичним працівникам, а також пацієнтам або їхнім законним представникам слід повідомляти про усі випадки підозрюваних побічних реакцій та відсутності ефективності препарату через автоматизовану інформаційну систему з фармаконагляду за посиланням: https://aisf.dec.gov.ua.
2 роки.
Зберігати в оригінальній упаковці для захисту від вологи. Не потребує особливих температурних умов. Зберігати у недоступному для дітей місці.
По 120 капсул у пляшці, по 1 пляшці у картонній коробці.
За рецептом.
Ф. Хоффманн-Ля Рош Лтд
Місцезнаходження виробника та адреса місця провадження його діяльності.
Віадуктштрассе 33, 4051 Базель, Швейцарія

Поки що відгуків немає
Будьте першими, хто розповість про свій досвід